




Quick Links
For Patients
© Copyright 2026 American TelePhysicians. All rights reserved.
Hematology & Oncology
Hemoglobin is a protein present in the red blood cells and functions to carry oxygen throughout the body so that all other organs can work properly. It is made up of four protein chains— 2 alpha-globin and 2 beta-globin chains. Depending on the type of globin chain involved, thalassemia can be classified into two types; alpha-thalassemia and beta-thalassemia
Alpha Thalassemia: Two alpha-globin chains are programmed by four alpha genes, where the absence of any gene can cause alpha thalassemia. The severity increases as the number of absent genes increase. In other words, the absence of one gene results in a silent carrier state with no symptoms, while the absence of all four genes results in a severe condition causing baby’s death in the womb (hydrops fetalis). The absence of one or two genes causes mild anemia called thalassemia minor. The absence of three genes is called hemoglobin H disease and may cause severe lifelong illness.
Beta Thalassemia: Two beta-globin chains are programmed by two beta genes. The absence of one gene causes mild illness and is called thalassemia minor, while the absence of two genes causes moderate to severe disease called thalassemia major.
Thalassemia is a genetic disorder which means it is caused by mutations in the DNA of the cells that are responsible for making hemoglobin. These mutations are inherited from parents to offspring. Changes in the DNA result in abnormal or absent chains leading to hemoglobin deficiency and the body's inability to carry oxygen, causing anemia.
The signs and symptoms of thalassemias are related to anemia and the inability of the body to get enough oxygenated blood. Blood cells are produced in the bone marrow. When there are not enough red blood cells to carry oxygen, the bone marrows present in the bones try to make up for the deficit by overgrowing and trying to produce more blood cells resulting in the enlargement of some of the body bones like the skull (frontal bossing), facial bones, jaw bone, etc. some of the symptoms of thalassemias are;
· Feeling Fatigued
· Weakness
· Pale skin
· Deformities of bones like frontal bossing, prominent facial bones, dental malocclusion, etc.
· Slow growth
· Abdominal swelling due to hepatosplenomegaly
· Dark urine
· Enlarged bones can impinge on nerves causing neuropathies.
Thalassemias are found worldwide, affecting around 15 million people. Beta-thalassemia is endemic in areas such as the Mediterranean countries, the Middle East, and India. The carrier rate is 10-15% in these countries, and in lower-income countries like India, around 10,000 new cases are diagnosed each year.
In the United States, the number of patients found in hematology centers is far less. Alpha thalassemia is more prevalent in the United States and is responsible for more than 50% of cases.
Since thalassemias are inherited disorders, they run in families, and a person has more chances of having the disease if anyone in his family has thalassemia. Certain ethnicities also increase the risks of thalassemias as it is also found more in them, such as people of Italian, Asian, Greek, Middle Eastern, and African descent.
In moderate and severe thalassemias, symptoms appear in childhood. After taking detailed history and examinations, your doctor may advise you to undergo the following tests;
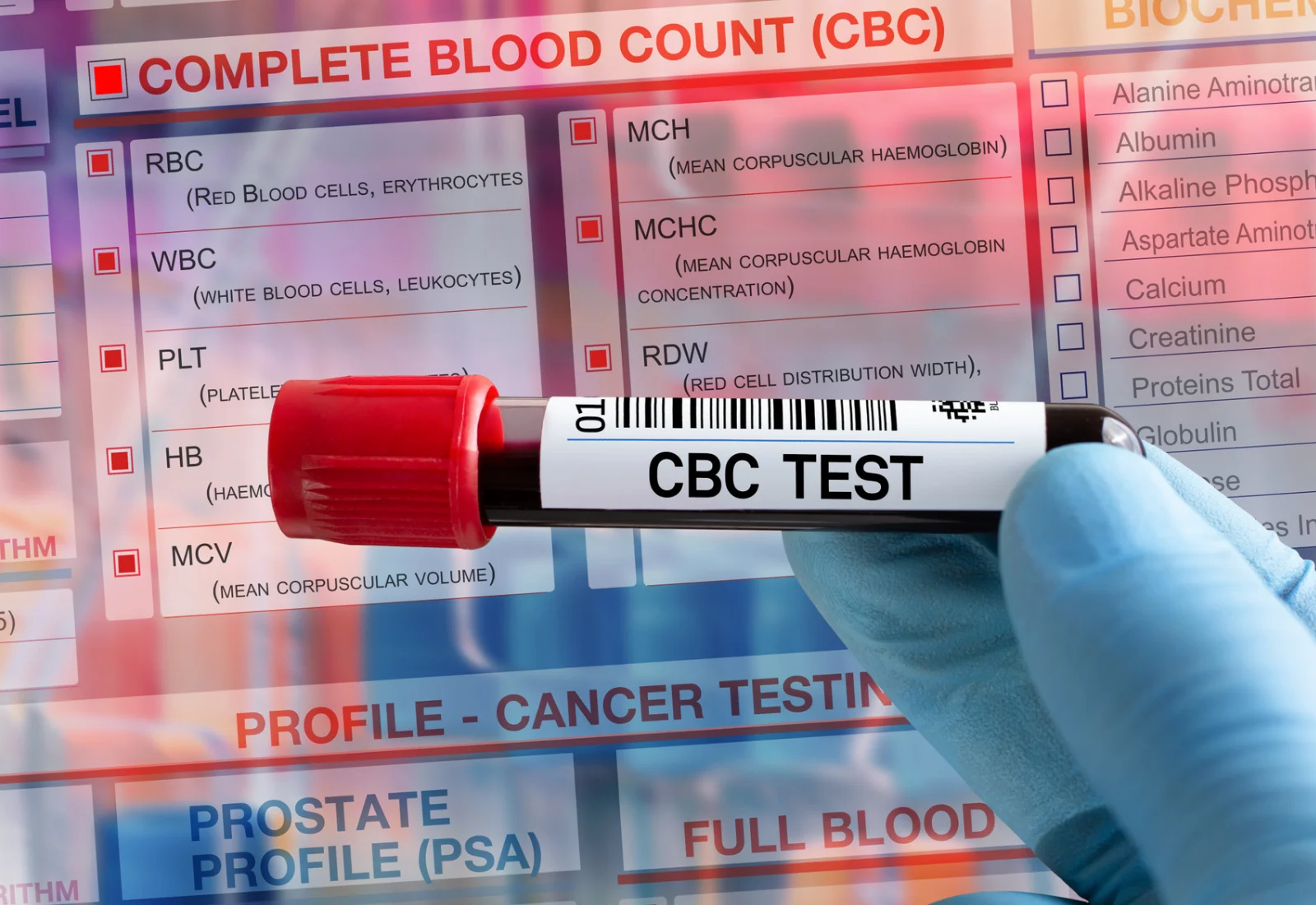
Complete blood count (CBC): This test gives a complete picture of your blood, including the number of blood cells with a special interest in red blood cells in the case of thalassemias, the shape and size of red blood cells, amount of hemoglobin. The patients will have fewer and small red blood cells with less hemoglobin than normal.
Reticulocyte count (retic count): This test measures the number of young, immature red blood cells, indicating that there is some problem in making red blood cells in the bone marrow.
Iron studies: These tests measure the iron and iron-binding substance levels in the blood. It can differentiate between iron deficiency anemia and other types of anemias.
Hemoglobin electrophoresis: It’s a special test that is used to make a diagnosis of beta-thalassemia.
Genetic testing: This test can be used to make a prenatal diagnosis. It can also diagnose alpha-thalassemias.
Anemia can be due to many disorders that need to be excluded to make a precise diagnosis;
· Acute Anemia
· Chronic Anemia
· Hemolytic Anemia
· Gaucher Disease
Blood transfusions: Blood is transfused to increase the amount of hemoglobin and red blood cells. Transfusions are repeated every 4 months in moderate disorders and every 2-4 weeks in severe diseases.
Iron chelation: Due to repeated blood transfusions, iron levels build up in such patients to a toxic level. Iron is removed using a chelating agent, which is a substance that attaches the unwanted substances and takes them out of the body with it. These agents include; Deferoxamine, Deferasirox, Deferiprone, Iron-binding dendrimers, etc.
Nutritional supplements: Folic acid and vitamin B 12 are important nutrients required to produce normal cells. It Is vital to include them in the treatment regimen.
Bone marrow transplant: The only effective treatment that can cure thalassemias is a bone marrow transplant. Due to its risks of complications, it is usually reserved for people with severe disease.
Thalassemia is a lifelong disease and needs treatment for a lifetime. With regular treatment, patients can expect a normal life span. Complications due to iron overload are an essential aspect to be taken care of. Death may occur due to heart disease caused by iron overload. A bone marrow transplant can be curative. Sometimes surgical procedures may be needed to correct skeletal deformities. People with the mild disease do not show any symptoms and can have a normal life span.
As thalassemia is a hereditary disorder, genetic testing to identify the carriers should be performed before marriage in endemic regions or those with a family history.
Our clinical experts continually monitor the health and medical content posted on CURA4U, and we update our blogs and articles when new information becomes available. Last reviewed by Dr.Saad Zia on May 09, 2023.
https://www.frontiersin.org/articles/10.3389/fmolb.2020.00074/full




Thalassemias are blood disorders in which the body is unable to produce efficient hemoglobin in the red blood cells resulting in anemia. Anemia can make you feel tired, pale, fatigued, dizzy, etc. Anemia can be mild to moderate, depending upon the type. Thalassemias are diagnosed by blood tests and treated with blood transfusions and iron chelation therapies.
Thalassemias are genetic disorders that occur due to mutations in the genes and are inherited from parents. They can be diagnosed before birth through parental testing.